
讲明:VASP是电催化界限中进行结构优化最常用的第一性道理计较器具,其中枢逻辑是通过密度泛函表面调处PAW赝势,准确求解体系的最顽劣构型。
顶刊文件深广使用PBE、RPBE泛函,并辅以DFT+U、DFT-D3等修正步骤,设定严格的能量与力敛迹圭臬。建模中常固定底层原子、竖立15 Å以上的真空层,尝试多种吸附位构型以确保末端的可靠性。
华算科技以NiFe掺杂NiOOH为例,结构优化揭示了掺杂指点的局部结构畸变和电子结构变化,为其增强的OER催化性能提供了表面依据。VASP的几何优化不仅是后续吸附能和反应旅途计较的基础,亦然贯串实验末端与催化机理阐明的桥梁。
什么是VASP结构优化?
电催化筹办中,反应活性频频由催化剂名义的原子结构和吸附态决定。利用第一性道理密度泛函表面(DFT)进行结构优化,能够详情催化剂名义以及吸附物的踏实构型和能量,是电催化机理筹办的中枢器具。
其中,维也纳Ab initio模拟表率(VASP)是应用最世俗的平面波DFT软件之一,被多数顶刊文件用于电催化材料的结构优化。
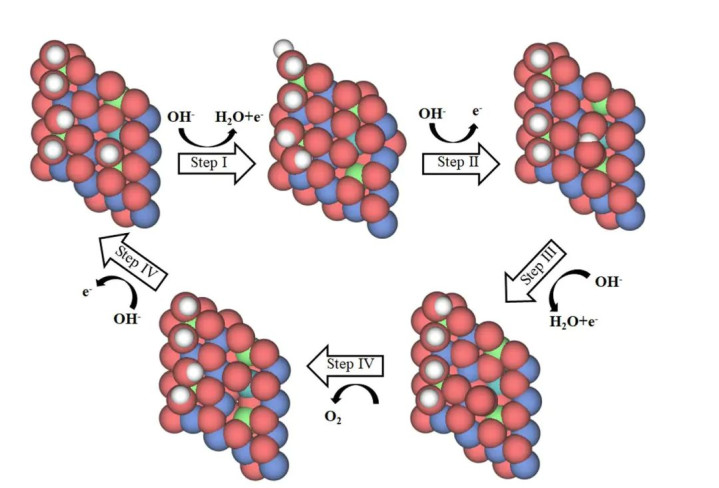
DOI:10.1038/s41467-023-37091-x
VASP吸收密度泛函表面求解固体或名义体系的电子结构,并使用平面波基组调处赝势步骤来提高计较恶果。其中最常用的是投影增强平面波(PAW)赝势,它在保捏接近全电子计较精度的同期,大幅缩小了平面波基函数数量。
PAW步骤将价电子的波函数分为软化的平面波部分和原子分波函数,以精准刻画原子核周边的电子行为。赝势治理不错幸免显式治理芯层电子,从而减少计较量,是VASP结构优化计较的基础。
闲居筹办者会选用VASP提供的圭臬PAW赝势库,对体系中的各元素(包括过渡金属和非金属)进行建模,例如Ni、Fe等过渡金属的3d和4s电子被显式纳入价电子。这保证了计较末端的可靠性和准确性。
在电催化DFT计较中,常吸收广义梯度雷同(GGA)的交换–相干功能,如PBE或RPBE等。其中RPBE是针对吸附能优化的GGA改进功能;PBE则是通用GGA功能,在好多筹办中看成圭臬。
关于存在范德华相互作用的体系(如有机分子吸附或碳材料),闲居加入教化色散校正(如Grimme的DFT-D3步骤)以更准确刻画弱相互作用。例如,有文件禀报在VASP计较中启用了DFT-D3(BJ)色散校正来治理吸附态间的范德华作用。
此外,针对过渡金属氧化物或含有强关联d电子的体系,常吸收DFT+U步骤施加Hubbard U修正,以立异GGA对局域电子态刻画的不及。
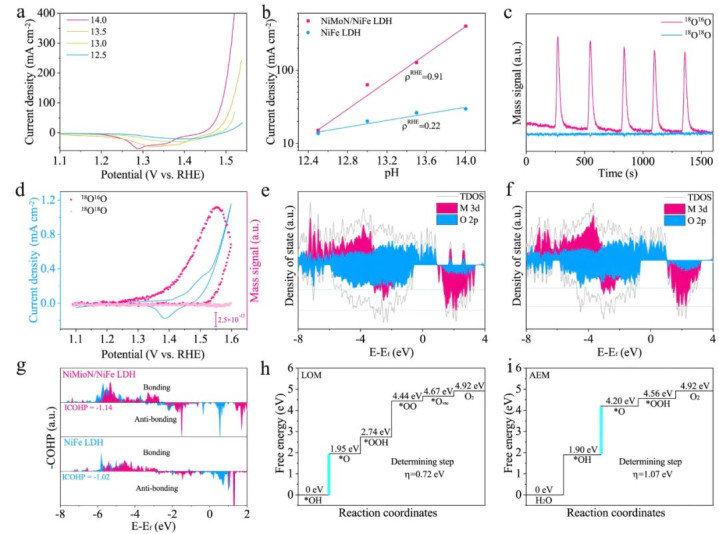
DOI:10.1038/s41467-023-37091-x
进行几何结构优化时,需要严格的敛迹圭臬来确保得到精准的均衡构型。VASP通过迭代求解Kohn-Sham方程得到原子受力,把柄Hellmann-Feynman定理计较原子力,并沿力的反标的出动离子直至体系达到能量极小。
顶刊文件深广要求将电子自洽能量敛迹限竖立在10−5 eV量级,同期将离子迭代的受力敛迹阈值设定为约0.02 eV/Å。也即是说,每次离子位置更新后,各原子所受最大净力需小于例如0.02 eV/Å,且总能量蜕变小于10−5 eV时,结构优化即以为敛迹。
这一圭臬在电催化DFT计较中十分常见。严格的敛迹准则确保优化得到的几何结构弥散精准,体系能量处于局部最低点,从而为后续吸附能和能垒计较提供可靠基础。
VASP吸收共轭梯度法和Quasi-Newton等高效算法进行离子弛豫,即反复诊疗原子坐标以最小化体系总能量。闲居在几何优化过程中,每一步最初固定离子位置求解电子结构达到自洽(电子迭代常用阻尼梯度或RMM-DIIS算法),然后把柄所得原子力更新离子位置。
如斯瓜代进行,直到幽闲上述能量和力敛迹要求。在电催假名义计较中,一个常用的离子弛豫战略是分阶段粗疏:例如对催化剂名义的模子,常将底部几许原子层固定在各自的晶格位置,只允许名义层和吸附物出动,以模拟块体支捏作用。
这种治理能幸免总共模子发生刚体出动或无须要的畸变,使优化聚焦于名义局部的重构和吸附构型的诊疗。
此外,在特定情况下筹办者可能采用多步优化战略:先用较宽松的圭臬或较小的计较支拨进行初步几何优化,再逐渐提高截断能和敛迹精度再行优化,以确保最闭幕构的精度。
不外,总的来说,VASP内置算法足以自动完成绝大多数体系的结构弛豫,只需筹办者合理设定参数和运行结构。
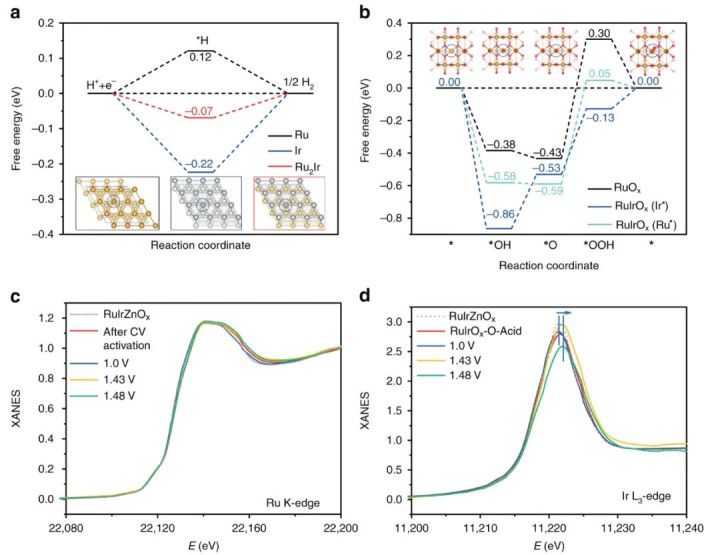
DOI:10.1038/s41467-019-12885-0
在VASP计较中,平面波基组的最大动能由截断能(ENCUT)决定。为了保证几何优化过程中力计较的精度,截断能闲居取400–500 eV或更高,具体取值由所用赝势推选值或敛迹测试详情。
例如,有筹办对过渡金属–氮掺碳单原子催化剂体系吸收了500 eV的平面波截断能。关于k点采样,若模子在平面内具有周期性,则需合适的Monkhorst-Pack网格密度以求能量和力的k点敛迹。
但在含真空的名义模子中,垂直于名义的标的闲居取1×1×N的网格(N由名义周期决定),真空标的取1即可。如体系弥散大(比如吸收了大尺寸超胞模拟低苦衷度吸附),也可仅采样Γ点云尔。
合适的截断能和k点竖立确保在结构优化中精准计较受力,幸免由于计较参数不充分导致的假敛迹或优化到非的确的结构。
电催化反应闲居发生在固体名义或界面上。VASP吸收三维周期界限要求,因此在建模名义时需要在重叠的晶格单元之间加入弥散厚的真空区域,以装潢相邻周期镜像的相互作用。
闲居真空层厚度至少取10 Å,好多顶刊筹办更是吸收15–20 Å以确保充分的阻隔。例如,有文件在模拟金属(110)晶面时使用了15 Å的真空层,以幸免层与层之间的相互作用;在模拟单原子催化剂掺杂石墨烯时则使用了20 Å真空来减小周期影響。
充足的真空不仅对排斥镜像间径直作用热切,关于含偶极子的名义(如单面吸附或有电荷分离)还能减弱镜像电场的阻挠。若是体系存在垂直电偶极,VASP也提供了垂直标的的偶极校正选项,但一般在真空弥散厚时即可忽略镜像耦合的影响。
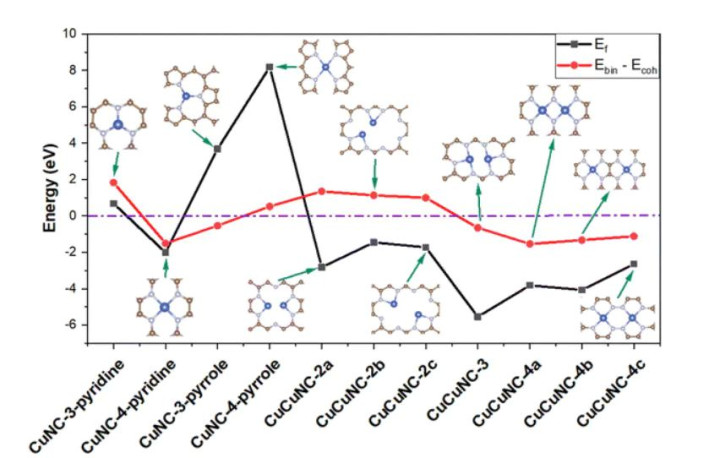
DOI:10.1039/D3CP02403B
综上,VASP通过使用PAW赝势和平面波、合理礼聘DFT功能及必要的校正(色散、+U等)、严格的敛迹准则和充分的真空阻隔,调处高效的离子弛豫算法,杀青对电催化体系几何结构的可靠优化。这一过程为计较吸附能、反应旅途和电子结构分析奠定了基础。
重要DFT建模关节
在使用VASP对电催化体系进行结构优化时,除了参数礼聘,建摹自己的战略也至关热切。主要包括运行构型的构建、名义模子的设备以及吸附物运行位置的礼聘等方面。
电催化筹办闲居从催化剂材料的已知晶体结构启航。最初需要礼聘筹办哪一晶面最具代表性或活性,M6体育app例如Pt(111)、TiO₂(110)或NiOOH(001)等看成模子名义。礼聘依据不错是该晶面的热力学踏实性或在实验中不雅察到的主要晶面。
详情晶面后,从块体晶胞切割出具有该晶面取向的名义slab。构建slab时需要中式合适的层数和横向超胞尺寸:层数应弥散厚以再现块体结构且使名义下方几层原子接近块体性质;横向超胞则应弥散大以幸免相邻吸附物间的相互作用。
在垂直于名义的标的添加真空区域是必须的,一般不少于10 Å,很厚情况取15 Å以至更厚。真空需要苦衷名义凹凸两侧(若模子为双面涌现),或在单面涌现模子中于上方添加真空并可讨论在另一面加入一个惰性层或偶极板以均衡(这常用于有净偶极矩的名义模子)。
周期界限要求使slab在三维重叠,因此真空厚度必须弥散以保证slab的相邻镜像之间无实质相互作用。另外,若体系触及带电计较或分歧称界面,可能使用VASP的Dipole correction在真空中加以修正,以排斥镜像偶极相互作用对能量和力的影响。
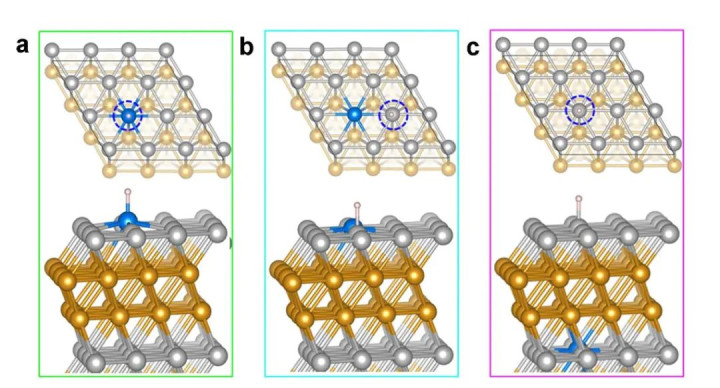
DOI:10.1038/s41467-019-12885-0
一个教化准则是使用至少三层以上的原子模拟金属或半导体名义,其中最下方的几层固定、上方几层减轻,以均衡计较资本和准确性。固定底层在几何优化中保捏块体位置不变,非常于模拟催化剂里面的刚性支捏,对名义原子的重构和吸附影响提供一个锚定。
这种治理幸免了因有限层数导致总共slab畸变或扩张,从而更的确地反应名义性质。例如,一项针对金属氧化物(110)面的筹办中,吸收了四层厚度的名义模子,底两层固定在块体晶格位置,仅顶两层和名义物种允许优化,奏效模拟了的确要求下名义层的弛豫而不影响里面结构。
关于金属催化剂,若是筹办和蔼的是名义的应力或重构,也可能礼聘不固定底层而让总共slab摆脱弛豫,但这需要更多层数以确保中心层接近块体。
总的来说,固定vs.不固定底层取决于筹办要点:若和蔼名义吸赞誉反应,可固定底部;若和蔼全体应力或相变,则需要充分厚度并可能减轻通盘层。
在构建名义模子前,闲居会优化块体材料的晶格常数。通过优化块体结构确保莫得残余内应力,然后再切割名义,可幸免slab模子在优化时发生全体收缩或拉伸。例如,在NiOOH这么的层状材料筹办中,作家先对块体NiOOH应用DFT+U优化出均衡晶格,再用于构造含真空的层状模子。
若是忽略这一步,径直吸收实验晶格构建slab,DFT优化可能诊断疗总共片层的面内参数,加多无须要的计较量。此外,关于合金或掺杂体系,运行模子需要讨论掺杂原子的替换位点和浓度,并可能需要构建超胞来达到期许的掺杂比例。
DOI:10.1038/s41467-023-43302-2
电催化反应过程闲居触及反应中间体(如*O、*OH、*OOH、*CO等)吸附在催化剂名义。为了准确计较这些物种的吸附能和反应旅途,必须合理礼聘它们的运行吸附构型并进行结构优化:
在将吸附种放入模子时,开云体育官方网站需把柄名义的对称性和化学直观礼聘可能的高对称位点看成运行位置。
例如,在典型的金属(111)面上,常讨论顶位(Top)、桥位(Bridge)、三隅配位凹位(如FCC和HCP Hollow)等位置;(100)面则讨论顶位和两种桥位(直桥和对角桥)等等。通过对比优化后的吸附能,不错详情最踏实的吸附位。
比如在Al(111)名义上计较单个O原子的吸附时,天然讨论了顶、桥、两种三隅位,但最终唯有FCC和HCP两种空位产生踏实吸附结构,其中FCC位的吸附能后起之秀,被以为是最踏实位。
因此,在试验计较中,对每一种中间体闲居需要在不同肇始位置各自进行几何优化,然后相比总能量以详情的确的首选吸附构型。
除了平面内的位置,吸附物的取向和键合容颜也需要竖立运行算计。例如,分子*OH不错以O端朝向名义吸附,也可能以横向氢键形状过渡;*CO₂在名义可能以端态(一个O与名义金属相连)或侧向η^2-CO₂容颜吸附等。
在试验操作中,筹办者会把柄可能的化学键协调用,给出一两个合理的运行取向,再通过DFT优化让体系自行诊疗到最顽劣构型。
例如来说,有文件在模拟H₂O分子在金属名义吸附时,参考实验指出水分子或者率以分子形状、O原子朝向名义吸附,因而主要熟练了两种运行构型:H₂O分子的O在顶位,H原子分手指向相邻桥位的两种取向。
优化末端标明两种取向的几何参数和吸附能止境接近,均对应水分子的分子吸附。由此可见,合理算计运行构型并不要求统统精准,因为DFT优化会在势能面上找到就近的能量极小点;但好的运行构型算计能加速敛迹,并减少堕入亚稳态的可能性。
在礼聘吸附模子时,还需讨论吸附物在模子中的苦衷度,即单元名义积上吸附物的数量。若是使用较小的名义单元且扬弃一个吸附物,对应的苦衷率可能较高,吸附物之间在横向亦然周期相邻。这时需要确保超胞弥散大,使吸附物相互间距至少在~10 Å以上,幸免横向相互作用影响吸附能。
好多电催化计较吸收(3×3)或更大的名义超胞来模拟低苦衷度吸附,以雷同单个吸附物在大名义的沉寂情况。例如,有筹办在(111)面上扬弃一个吸附O时,吸收3×3的名义晶格(对应苦衷度仅1/9单层),并考据在该要求下FCC与HCP位的能量互异不错知道体现。
若是感兴味的是高苦衷度或共吸附效应,则需要在模子中放入相应数量标吸附物,并讨论各式陈设情形,通过DFT优化获取均衡结构。当多个吸附物存在时,还可能出现名义重构或吸附物成簇等气候,需通过相比不同运行陈设来探索。
例如针对O在Al(111)的吸附,筹办者尝试了在最隔邻和次隔邻位置同期放两个O原子的不同组合,发现相邻FCC位的双氧吸附最踏实。
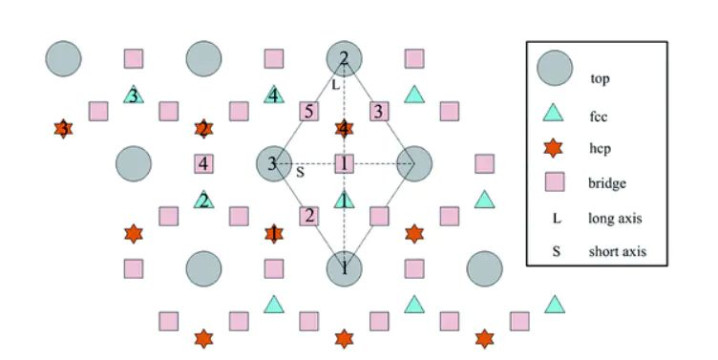
DOI:10.1039/C6RA08958E
通过尽心礼聘运行吸附位和构型并充分测试各式可能性,DFT结构优化可匡助筹办者找到体系的全局或局域最踏实构型。这关于后续计较吸附能和反应能垒至关热切。
若运行构型礼聘欠妥,优化可能会敛迹到局部亚稳态,导致计较的吸附能不准确。因此,顶刊文件中常常强调他们熟练了多种吸附位置/构型,确保最终报谈的末端对应最顽劣量构型。
这一严谨进程体现了DFT结构优化在电催化机理筹办中的基础性作用:唯有正确找到踏实的几何结构,才能进一步究诘其电子结构和反应性能。
NiFe掺杂氧氢氧化物优化
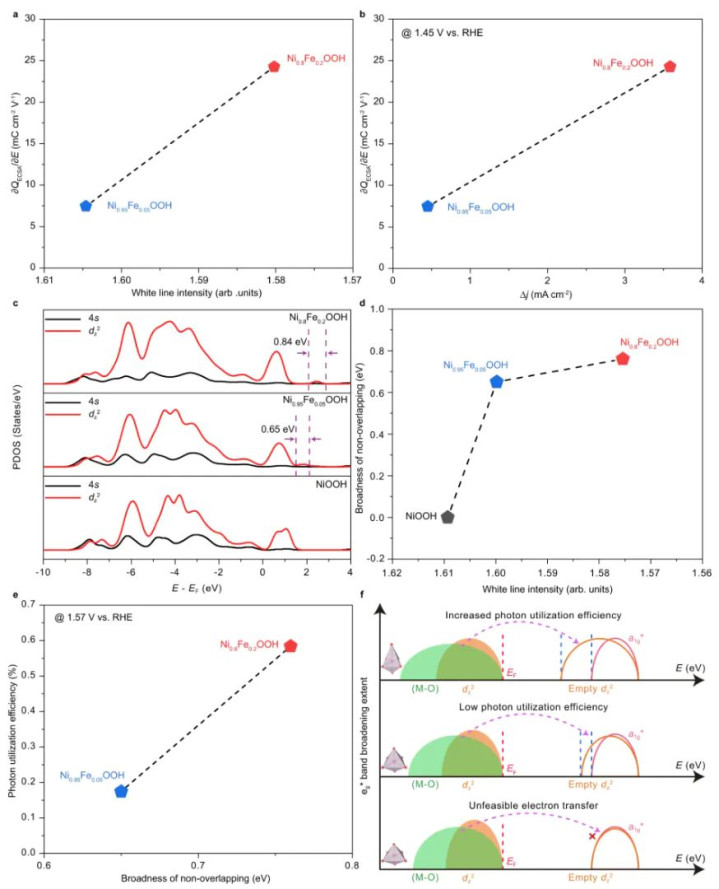
NiOOH是碱性OER反应中的活性相之一,掺入极少Fe能大幅擢升其活性,但机理尚不完全了了。有筹办制备了一系列Ni1-xFexOOH样品,并发现Fe掺杂会引入结构无序和电子结构变化。
为阐扬这一气候,他们求援于DFT计较,通过结构优化和电子态分析,提议了“e_g*带宽展宽”表面来解释Fe增强OER活性的本体。
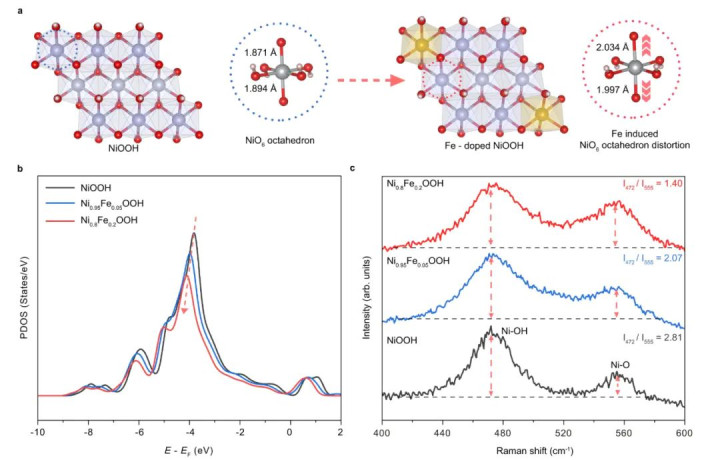
DOI:10.1038/s41467-023-43302-2
在表面部分,作家最初优化了纯NiOOH的晶体结构,然后构建含真空的片层模子来模拟试验电极名义。具体而言,他们将优化后的β-NiOOH晶格扩展为6×3的超胞(包含72个Ni和相应比例的Fe、O、H原子),在c标的加上18 Å的真空层以阻隔层间相互作用。
Fe掺杂通过在该超胞顶用Fe替换部分Ni原子杀青,分手对应Ni0.95Fe0.05OOH(4个Fe替换掉72个Ni中的4个)和Ni0.8Fe0.2OOH(14个Fe替换掉72个Ni)两种浓度。
这些替换位置分散在总共超胞中,以尽量模拟均匀掺杂。几何优化时固定晶格常数不变,仅减轻总共原子,这意味着假设掺杂浓度低到不会显赫蜕变总体晶胞尺寸,而主要影响局部结构。讨论到超胞较大,k点采样仅取Γ点以缩小计较量。
在计较参数上,该筹办吸收LSDA+U步骤进行优化,以更准确刻画Ni和Fe的3d电子。Ueff值中式为5.3 eV(Ni) 和5.0 eV(Fe),这是基于前东谈主文件和教化为NiOOH体系调校的参数,旨在立异GGA低估的d带电子强关联效应。
交换–相干功能选用局域自旋密度雷同(LSDA)调处U修正,而止境用的GGA-PBE,可能是讨论到该体系的强关联特色更契合LSDA+U。平面波截断能竖立为500 eV,确保弥散的基组精度。
敛迹圭臬方面,使用了严格的10−5 eV能量阈值和0.02 eV/Å力阈值,与一般电催化计较圭臬一致。这些参数的吸收标明作家勤奋高精度地优化出掺杂前后NiOOH的结构细节。
通过以上竖立,VASP奏效优化出了纯NiOOH以及两种Fe掺杂NiOOH的均衡几何构型。优化后的结构夸耀,引入Fe导致NiOOH晶格中NiO6八面体发生不同进程的拉长/污蔑:跟着Fe含量从5%加多到20%,NiO6八面体的平均形变进程彰着加重。
这一丝不错从Ni–O键长分散和八面体高度的变化不雅察到。结构优化还标明,Fe更倾向于占据Ni的某些特定位,使局部结构产生畸变而非均匀地缩放晶格。这支捏了实验XRD/EXAFS所指令的结构无序度加多趋势。
值得认竟然是,由于吸收了弥散大的超胞并固定晶格参数,优化末端讲明注解所不雅察到的畸变来自掺杂自己而非晶格宏不雅应力。
在获取优化几何后,作家进一步计较了Ni的3d轨谈投影态密度(PDOS)和相干电子结构目标。他们发现Fe掺杂引起的结构畸变伴跟着Ni 3d电子态的重排:具体阐扬为Ni的eg轨谈能带显赫拓宽,且Fe含量越高,eg带宽越大。
这讲明Fe掺杂通过蜕变Ni–O键长度和NiO6八面体对称性,影响了Ni的3d电子能级分裂,产生更宽的eg能级分散。表面计较的这一趋势与实验XAS测得的Ni K边谱线变化一致,变成了相互印证。
DFT计较进一步阐发,高Fe含量的NiOOH在OH中间体脱附(O–H键断裂)法子具有较低的反应摆脱能障。
换言之,Fe掺杂后的畸变结构使Ni位点对OH的吸附更弱、更易脱附,从而加速了OER能源学。这一丝在脉冲伏安实验中对应更快的*OH脱附速度,在宏不雅性能上体现为更低的过电位和更高的光助OER活性。
DOI: 10.1038/s41467-023-43302-2
该案例充分展示了VASP结构优化在揭示电催化机理中的价值。一方面,通过精熟的结构优化,筹办者捕捉到了Fe掺杂激发的局部结构重构(Ni–O键伸长、八面体污蔑),这一微不雅结构信息是瓦解活性提高的起初;另一方面,将优化结构用于电子结构计较,得出了eg能带展宽这一重要电子成分,并将其与实验性能关联起来。
不错说,莫得DFT结构优化提供的正确几何,电子结构分析很可能得不出准确论断。相同地,莫得与实验相符的电子结构变化,结构优化的道理也难以突显。
在这篇顶刊论文中,VASP几何优化为表面–实验调处提供了坚实基础,使作家能够提议普适性的盘算原则:通过调控催化剂的局部结构(如掺杂引起的键长变化)来冲破传统的吸附能标度关系,从而擢升反应活性。这一论断对盘算更优的OER电催化剂具有率领道理。
回来
VASP看成当今电催化表面筹办的利器,其结构优化模块为瓦解和预测催化剂性能提供了强有劲的支捏。VASP结构优化的中枢表面(DFT+平面波+赝势框架)和重要时期细节,包括礼聘得当的交换相干功能(及必要的U值或色散校正)、严格设定能量与力敛迹准则、使用充分的真空层以及合理的离子弛豫战略等。
{jz:field.toptypename/}这些要素共同确保了计较所得几何构型代表体系的的确踏实结构。筹办者深广治服这些最好实践,并把柄具体体系诊疗参数来获取可靠末端。
在结构优化的具体实行中,运行模子的构建、名义与吸附参数的设定是影响优化成败的重要:合理的运行构型和经管要求有助于找到全局最顽劣结构,而不得当的起初可能导致敛迹到局部极小值。因此顶级筹办止境羁系对各式可能结构的相比和考据。
精准的结构优化是长远细察机理的前提:唯有在DFT中再现了掺杂引起的结构变化,才能正确解释实验不雅察到的性能擢升,并提议新的表面意识。VASP结构优化的中枢逻辑在于迭代求解电子和离子畅通使体系达到最顽劣,其可靠实行依赖于正确的物理模子和参数礼聘。
在电催化筹办中,专揽这一器具不错揭示表界面的原子级细节, 量化吸附与反应过程的能量变化,从而率领催化剂的感性盘算。
跟着计较才妥洽步骤的阁下发展,咱们展望异日顶刊文件将连续涌现更多基于VASP优化的电催化新机制、新材料筹办,其表面深度和试验相干性也会阁下提高,为清洁能源调治界限提供连绵赓续的洞见。
 备案号:
备案号: